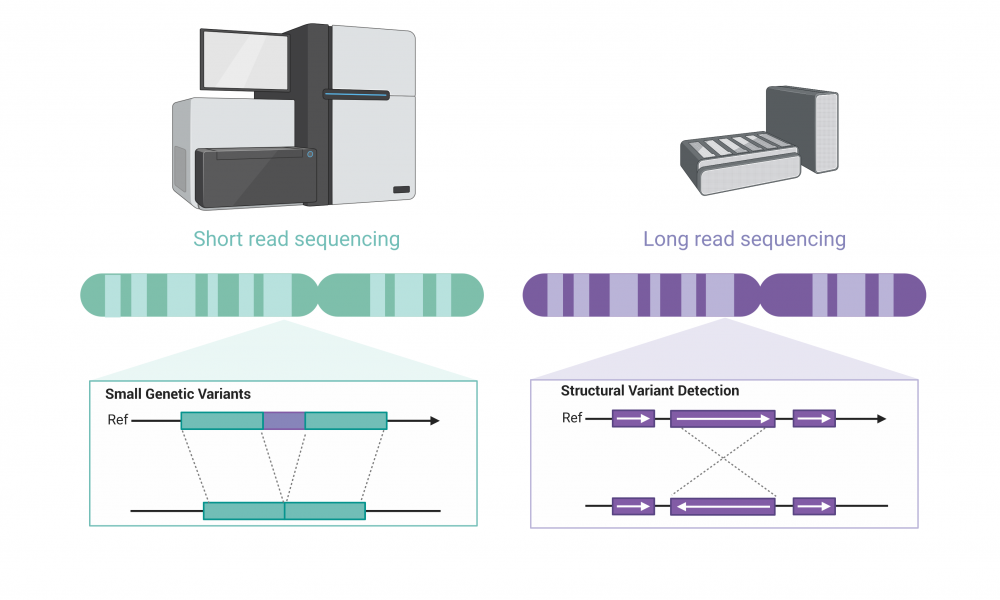
Whole genome sequencing has revolutionized cancer treatment planning, enabling the rapid detection of DNA mutations not routinely screened for in the clinic. Until recently, however, scientists were limited in their ability to detect large structural genetic variants using short-read sequencing technology. But with the recent release of long-read sequencing instruments, cancer researchers have been eager to see what such technology could mean for precision oncology.
Now, a new study published in Genetics in Medicine led by Dr. Steven Jones, Co-Director and Head of Bioinformatics at the GSC and Dr. Kasmintan Schrader, Scientist, Staff Physician and Co-Director of the Hereditary Cancer Program (HCP) at BC Cancer, illustrates the significant benefit of implementing long-read sequencing into precision oncology.
In this study, researchers employed the Oxford Nanopore PromethION (a long-read sequencer recently added to the armament of instruments at the GSC’s technology platform), to re-sequence and analyze the genomes of cancer patients enrolled in BC Cancer’s Personalized OncoGenomics (POG) program, which were previously sequenced using only short-read technology.
“We wanted to show that long-read sequencing, which is a fairly recent technology and is not yet implemented clinically, is complementary to other sequencing technologies that are used more commonly in clinical and research settings,” says Katie Dixon, graduate student in Dr. Schrader’s group and co-first author on the study.
Short-read sequencing
Since the advent of next generation sequencing technologies, DNA sequencers have rapidly evolved to accurately and efficiently sequence billions of bases of DNA. Next generation sequencing relies on short-read technology, meaning that the DNA is first sheared into short fragments and then sequenced—in the range of 150 to 250 bases—which are then assembled into the correct sequence and often aligned to a reference genome.
The high accuracy of short-read sequencing enables the identification of small genetic variations that may influence cancer progression and treatment response; however, the assembly of short reads poses a challenge when seeking to identify larger structural variants in the genome, such as inversions, translocations, insertions or deletions.
To help parse out these important genetic events that may have clinical implications, researchers are turning to long-read sequencing. The technology has lower accuracy on average in identifying individual bases and is less efficient at detecting small variants, but it is ideally suited for the identification of large changes in genome structure.
Long-read sequencing
Long-read sequencing instruments developed by Oxford Nanopore Technologies detect changes in an ionic current as a string of nucleic acid passes through a protein pore embedded within a membrane. These changes in current are then translated into a sequence of bases, enabling the production of DNA sequences of more than a million bases in length.
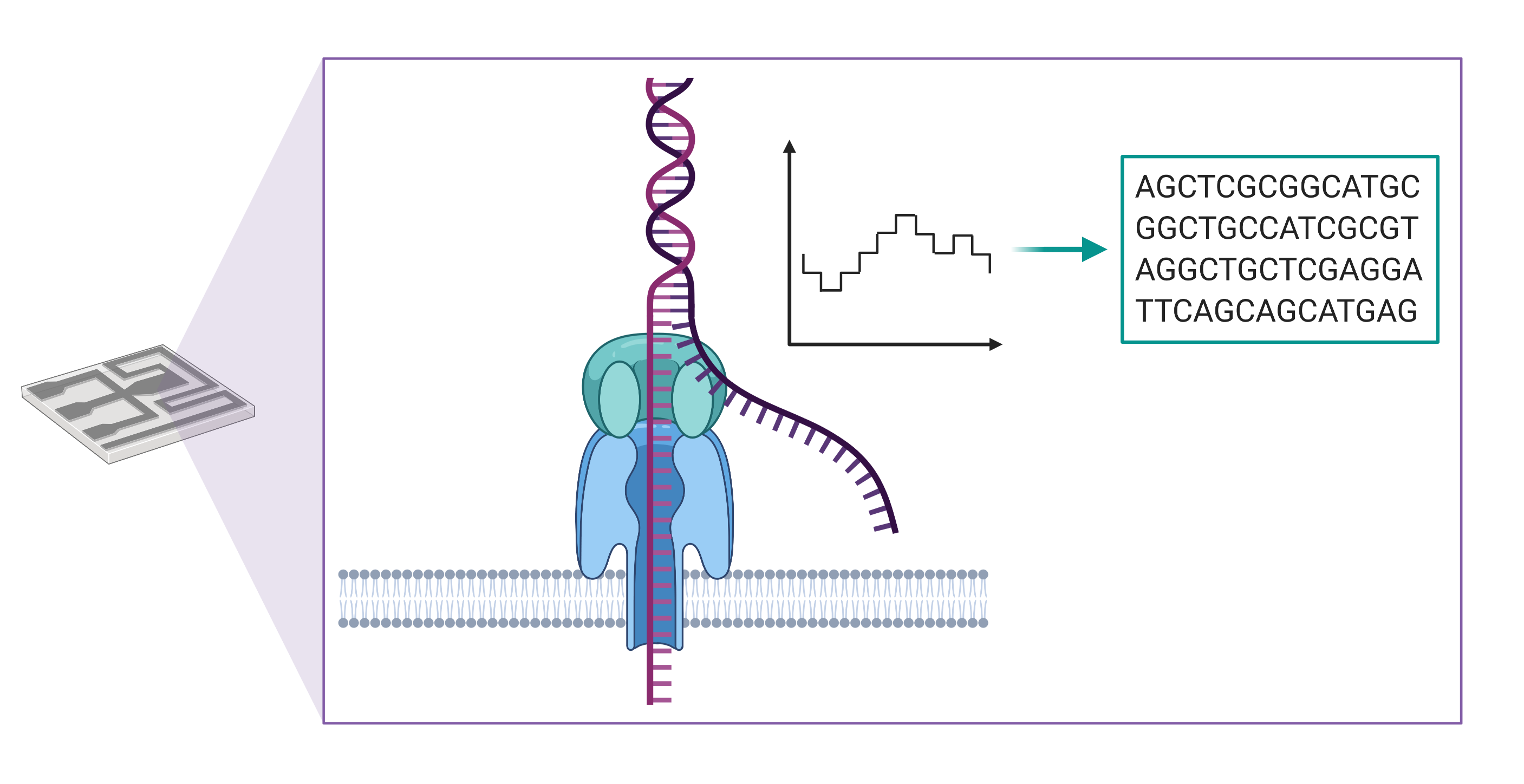
There are several advantages to using long-read sequencing compared with next generation sequencing. Longer reads allow for better overall resolution of highly repetitive genomic sequences, facilitating the assembly of large and complex genomes. For this reason, when aligning sequenced bases to a reference genome, long-reads better enable detection of large-scale changes in genomic structure, or what genome scientists refer to as structural variants. The technology also allows for the direct detection of epigenetic modifications, such as DNA methylation, which would require additional library construction and sequencing runs using short-read sequencing methods.
Long-read sequencing and precision oncology
The Genetics in Medicine study focused on germline variants—those that are inherited and shared among family members—which may influence cancer susceptibility, formation, progression and treatment outcomes. When germline variants are detected as part of the POG program, patients are referred to the HCP for genetic counselling and carrier testing for family members.
Out of the 669 patients within the cohort, 13 of 14 individuals found to have potentially damaging germline structural variants by short-read sequencing were analyzed by long-read technology. Nanopore whole genome sequencing confirmed eight variants that were detected by short-read sequencing and were likely to be involved in disease, and definitively resolved three additional variants that could not be fully characterized with short reads. This included reclassification of a variant detected in multiple patients as harmless, obviating the need for genetic counselling and clinical follow up.
“Re-sequencing using long-reads to confirm the findings from short-reads revealed that three patients had this false positive, which was completely opaque to short-read sequencing,” says Dr. Kieran O’Neill, Staff Scientist working under Dr. Jones at the GSC and co-first author on the study. “We were able to definitively say that it was not a harmful variant, which saved time and worry for patients and their families. We also detected events that were suspected by short-read sequencing but could not be verified, which we were able to elucidate using Nanopore long-read sequencing.”
Benefit to patients
Research has shown that germline variants in cancer predisposition genes underlie five to 10 per cent of all cancers. But many patients who appear to have a hereditary predisposition to cancer cannot be provided with a genetic explanation using standard clinical assays. The findings of this study suggest that, for these patients, long-read sequencing may be advantageous to confirm the presence or absence of germline structural variants that may have implications for their disease.
“There are a lot of individuals who come into the HCP who look like they could be suspicious for a genetic predisposition based on their personal and family cancer history, but after going through clinical testing they remain without a genetic diagnosis,” says Dr. My Linh Thibodoeau, Clinician Scientist at BC Cancer and co-first author on the study. “Long read sequencing may be able to fill in that gap for patients where nothing has been found using other methods.”
“This study highlights the utility of long-read sequencing in clarifying the genetic diagnosis associated with structural variation. It paves the way for consideration of this technology as an orthogonal method of genetic assessment in the clinic when validation or clarification of structural events is required, or for when no diagnosis is achieved through standard clinical testing,” says Dr. Schrader.
Implementation of long-read sequencing into POG
Integration of long-read sequencing into a program like POG requires technology development and optimization. As part of this study, the group developed processes for aligning long-read sequencing data and for detecting variants. Moving forward, further development is needed to allow for the detection of variants without prior knowledge.
“What we would like to do in the future is use our tool called MAVIS (Merging, Annotation, Validation and Illustration of Structural variants), which was developed at the GSC for detection of structural variants from short-read sequencing,” says Dr. O’Neill. “We would like to extend MAVIS to take in long reads and integrate all that information together to verify those structural variants.”
For now, the group is looking to employ long-read sequencing to further explore and validate structural variants detected by short-read sequencing as part of the POG program, using MAVIS to combine the two technologies in order to better inform patient care.
“We will use this technology to help diagnose families that are strongly suspicious for hereditary cancer syndromes who have not been diagnosed by current clinical sequencing technologies. Enhanced resolution and identification of germline structural variants will improve the diagnosis and management of hereditary cancer families, not only at a patient and family level, but also at a population level by informing the design of standard clinical assays,” says Dr. Schrader.
Learn more about Dr. Jones' research
Learn more about Dr. Schader's research
Learn more about the GSC's technology platform and software development
Learn more about the Personalized OncoGenomics Program
Learn more about the Hereditary Cancer Program
