
At present, multiple tools are available to assemble draft genomes using long reads and, separately, to correct errors. In a recent innovative development from the GSC’s Dr. Inanç Birol Lab, researchers have created LongStitch, a scalable pipeline that contains multiple tools (including Tigmint-long, ntLink, and ARKS-long) customized specifically for long read sequences to correct and scaffold draft genome assemblies.
Genomes from most species are long and complicated molecular structures comprised of DNA. The human genome, for example, is more than 3 billion base pairs long. Genomes contain the instructions for building living organisms that, when decoded, provide valuable information about the fundamentals of life, evolution, health, and disease processes.
In order to decode such a long, complex, and often repetitive molecule, the genome can be broken down into smaller fragments known as “reads” that can then be “stitched” together and analyzed as a whole: this process is known as de novo genome assembly. Sequencing reads can come in two forms: short or long reads.
Short-read versus long-read sequencing
Short-read sequencing technologies, also referred to as next generation sequencing (NGS) or second-generation sequencing, rely on fragmenting a long stretch of DNA (e.g., a genome) into short fragments (75-400 bp), each of which is then amplified and sequenced in parallel, ultimately saving time.
On the other hand, long-read sequencing technology, also referred to as third-generation sequencing, works with DNA that has been fragmented into much longer pieces (>10,000 bp). Unlike short reads, long reads are particularly valuable for providing sequencing information on repetitive genome regions. At present, one of the main drawbacks of long-read sequencing, however, is a higher error rate compared to short reads, though this is continually being improved upon.
De novo genome assembly scaffolding tools for long read sequences
Multiple bioinformatics tools, known as scaffolders, are presently available for improving draft genomes (without the use of a reference genome) using long reads (e.g., npScarf, OPERA-LG, LRScaf, etc.). However, many of these tools, while very adept at generating a contiguous genome, still contain errors.
GSC researchers from Dr. Inanç Birol’s Lab noted that such assemblies could benefit with a preliminary correction step prior to scaffolding. Therefore, to streamline current de novo genome assembly pipelines used for long reads, the researchers developed LongStitch.
According to one of the lead authors of the study, Lauren Coombe, assistant bioinformatics coordinator: “Generating genome assemblies is an essential step in many different genomics analysis pipelines, so it is very important to have tools which can use the rich information in sequencing data, particularly long reads, to achieve the highest quality genome assemblies.”
LongStitch for long reads
LongStitch is a genomic data processing pipeline that contains a variety of bioinformatic tools—all developed by the Birol Bioinformatics Technology Lab—and modified to work with long reads, specifically.
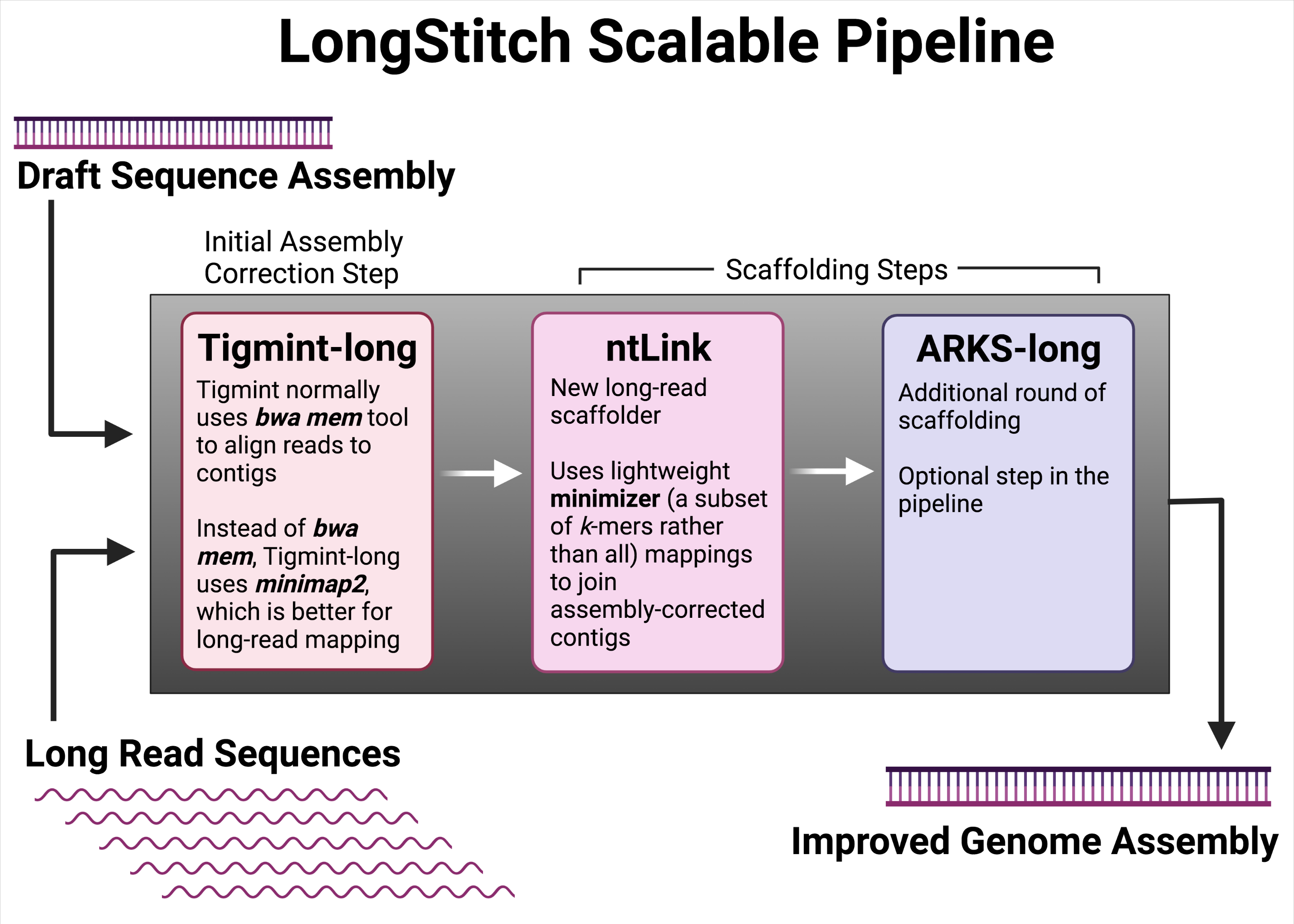
The researchers tested LongStitch using draft genome assemblies of various model organisms (C. elegans, O. sativa) and humans. They also compared their pipeline to the current gold standard long-read scaffolder, LRScaf, and found that LongStitch generated more contiguous and accurate assemblies in most tests.
In the future, other de novo genome assembly projects using long reads (e.g., sequencing the genome of newly discovered organisms) could benefit by using LongStitch.
“A huge benefit of using LongStitch is that it performs correction on the assembly as well as scaffolding, a combination not seen in the majority of other similar utilities,” said Lauren.
“Therefore, the assemblies output from LongStitch are more correct than existing long-read scaffolding tools in addition to increasing the sequence contiguity, which is very valuable to downstream analyses. Furthermore, LongStitch is fast and memory-efficient, making it an accessible pipeline for many different research groups.”
Acknowledgements:
This study was supported by funding from the National Institutes of Health, Genome British Columbia, and Genome Canada.
Image created with BioRender.com.
Learn more:
Learn more about the Birol Lab at the GSC and about their bioinformatics technology research
Learn about other bioinformatics tools and software for analyzing sequencing data, including the Birol’s Lab’s previously published tools, Tigmint and ARKS
Citation:
Lauren Coombe†, Janet X. Li†, Theodora Lo†, Johnathan Wong, Vladimir Nikolic, René L. Warren, Inanç Birol. LongStitch: high‐quality genome assembly correction and scaffolding using long reads. BMC Bioinformatics.
*bold font indicates members of the GSC.
† indicates authors that contributed equally to this work
