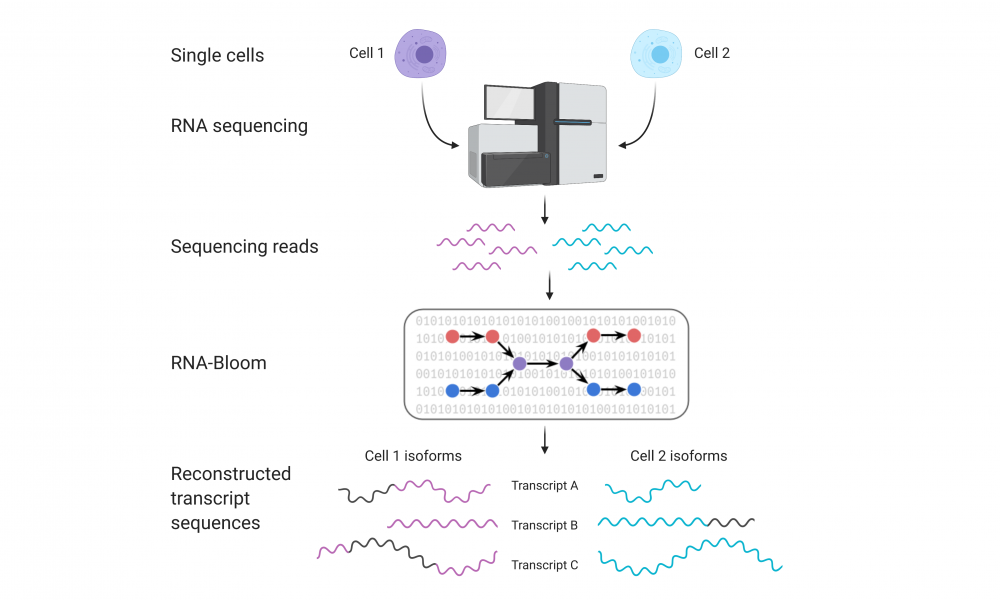
The advent of single-cell RNA sequencing technologies has provided unprecedented opportunities for the analysis of transcriptomes at single-cell resolution, allowing researchers to explore cell-to-cell variability. Now, researchers have developed a tool for the analysis and identification of RNA isoforms from single-cell RNA sequencing data.
Methodologies for the analysis of single-cell transcriptome data to date have primarily focused on gene expression, but analysis and identification of RNA isoforms—splice variants of transcripts from the same gene—and foreign transcripts from single-cell sequencing data has remained a significant challenge.
RNA isoforms arise from alternative splicing, a tightly regulated biological process that can greatly expand the number of protein products produced from a given gene. Deregulation of alternative splicing can disrupt important physiological protein functions and has been implicated in diseases including cancer, neurodegenerative disorders, cardiovascular diseases, metabolic conditions, and infectious and immune diseases.

In a new study published in Genome Research, GSC Distinguished Scientist Dr. Inanc Birol and his group describe a new tool, RNA-Bloom, that enables these analyses, providing a new avenue for the exploration of cell heterogeneity in alternative splicing and its involvement in disease.
“Typically, when scientists analyze single-cell RNA sequencing data they focus on gene expression profiles,” says Ka Ming Nip, PhD Candidate in Dr. Birol’s group and first author on the study. “But in this paper, we are providing the scientific community with a tool to look at single-cell RNA sequencing data from a different perspective. Instead of looking at the data at the gene level, RNA-Bloom enables researchers to reconstruct RNA isoforms at single-cell resolution.”
The release of RNA-Bloom from Dr. Birol’s group comes ten years after their Nature Methods paper describing Trans-ABySS, a widely employed bioinformatic tool for the assembly of RNA isoforms from bulk transcriptome data.
“Trans-ABySS was instrumental in many landmark cancer genomics studies. Now with RNA-Bloom, we are again advancing the field substantially,” says Dr. Birol. “The new tool not only improves upon transcriptome assembly algorithms, but also allows for studying transcriptomes at a single-cell level.”
While Trans-ABySS only allows reference-free assembly, RNA-Bloom can be either reference-free or reference-guided. In their study, the group compared RNA-Bloom to existing tools or RNA isoform assembly and found that, when compared to state-of-the-art reference-free and reference-guided tools, RNA-Bloom out-performed existing tools.
“The biggest challenge behind single-cell RNA sequencing data is something called drop-out, which leads missing data from individual cells. Typical approaches try to assemble the isoform sequences for individual cells but because of the missing data, it doesn’t matter how you tune the parameters, you can only go as far as the program was designed to do,” says Nip. “Using RNA-Bloom, we combine data from multiple cells and by doing so, we are able to recover the missing data from individual cells. We demonstrated that by using this method, we were able to construct significantly more isoform sequences than existing methods.”
Dr. Birol’s group has made RNA-Bloom freely available on GitHub.
The study: Ka Ming Nip, Readman Chiu, Chen Yang, Justin Chu, Hamid Mohamadi, Rene Warren and Inanc Birol. 2020. RNA-Bloom enables reference-free and reference-guided sequencing assembly for single-cell transcriptomes. Genome Research.
This work was supported by Genome Canada and Genome British Columbia, the National Institutes of Health and the Natural Sciences and Engineering Research Council of Canada.
Learn more about Dr. Birol’s research
Learn more about software development at the GSC
Images created using Biorender.com
